海南大学Small研究论文(芦俊达博士一作):构建钴镍双原子位点作为海水电池具有很强Cl-腐蚀性能的亲氧ORR催化剂
发布时间:2025-01-23
海南大学Small研究论文(芦俊达博士一作):构建钴镍双原子位点作为海水电池具有很强Cl-腐蚀性能的亲氧ORR催化剂

构建钴镍双原子位点作为海水电池具有很强Cl-腐蚀性能的亲氧ORR催化剂
第一作者:芦俊达
通讯作者:郑学荣教授 王杨副教授 邓意达教授
单位:海南大学
链接:https://doi.org/10.1002/smll.202407339
研究背景
海洋经济的蓬勃发展和对海洋资源开发的追求刺激了电子设备在海水中的应用。为了保证这些设备的长期运行,需要有可靠的电源。海水电池(SWBs)成为一种很有前途的候选电池,它通过消耗海水中的溶解氧(DO)来工作(它也被称为溶解氧海水电池)。与水下设备中常用的锂离子电池和铅酸电池相比,SWB具有高度内在的安全性、低成本、高能量密度、长期稳定性和环境友好性。然而,SWBs的性能高度依赖于阴极电催化剂,因为ORR动力学缓慢,经历了复杂的多步质子耦合过程此外,在恶劣的海底环境中普遍存在的极低的DO浓度(约为0-9.6 mg·L−1),进一步限制了阴极ORR速率和电池性能。此外,Cl−在海水中的阻断和腐蚀作用极大地影响了ORR电催化剂的活性和长期稳定性,因为Cl−优先吸附在电催化剂的表面催化位点,从而中毒了活性位点。因此,开发高效稳定、高亲氧性吸附氧分子和在海水电解质中耐Cl−腐蚀的阴极ORR催化剂势在必行。虽然铂基电催化剂表现出良好的ORR性能,但其昂贵、稀缺性、容易在海水电解质中的失活和腐蚀阻碍了它们的广泛应用。单原子催化剂(SACs),主要是金属氮-碳(M-N-C、M-Mn、Fe、Co等)。具有M-N4活性位点的材料,由于其高效的原子利用和灵活的电子结构调制,具有取代铂基电催化剂的巨大潜力。然而,单催化位点仅与O2分子具有特定的吸附和偶联模式,其中O2通常通过端点模式垂直吸附到M-N4平面。然而,在这种模式下,很难打破线性缩放关系以显着优化 ORR 中间体在单个金属位点上的吸附能量。最近的研究表明,双原子催化剂(DACs)具有克服上述限制的潜力,可以实现快速的四电子转移途径,并通过解离过程促进ORR动力学,从而优化O2的吸附行为。当O2被桥模型吸附时,更容易发生O-O键分裂为*O(*为活性位点),并跳过基于端子模型的结合途径(*O2→*OOH)的缓慢动力学。当O2被桥模型吸附时,更容易发生O-O键分裂为*O(*为活性位点),并跳过基于端子模型的结合途径(*O2→*OOH)的缓慢动力学。合理构建相邻金属位点可以调节O2的吸附行为,提高其亲氧性和吸附模式,加快海水中的ORR动力学。然而,精确设计相邻双原子活性位点的方法和阐明ORR的电催化机制仍然是一个重大挑战。此外,对Cl−的抗性也是提出DACs在海水电解质中应用的关键。然而,设计具有高活性和抗Cl−腐蚀的DACs,以提高其在海水环境中的催化活性和稳定性,促进其在污水处理厂中的应用是一个巨大的挑战。
文章简介
近期,海南大学郑学荣教授、王杨副教授、邓意达教授等合作在Small发表了题为“Building Cobalt-Nickel Diatomic Sites as Oxygenophilic ORR Catalyst with Strong Cl−-Corrosion Resistance for Seawater Batteries”的研究论文。该研究提出了一种主客体策略,在氮掺杂碳(CoNi-DAC)上制备具有相邻Co和Ni位点的双原子催化剂,其中Co和Ni原子分别配位到三个氮原子上。理论计算和原位表征表明,Co和Ni价态的同步还原通过优化O2吸附能垒,促进了O-O键的直接裂解,防止了*OOH中间体的形成,从而增强了ORR动力学。这种电子调制增强了亲氧性和Cl−的耐腐蚀性。Co/Ni双原子位点协同提高了ORR的催化活性,达到了0.79 V的半波电位(E1/2)和在天然海水中近700 h的特殊长期耐久性。用CoNi-DAC涂层碳刷电极组装的SWB的峰值功率密度为3.3W·L−1。这项工作为自然海水环境中先进的ORR电催化剂的设计和开发提供了有价值的见解。
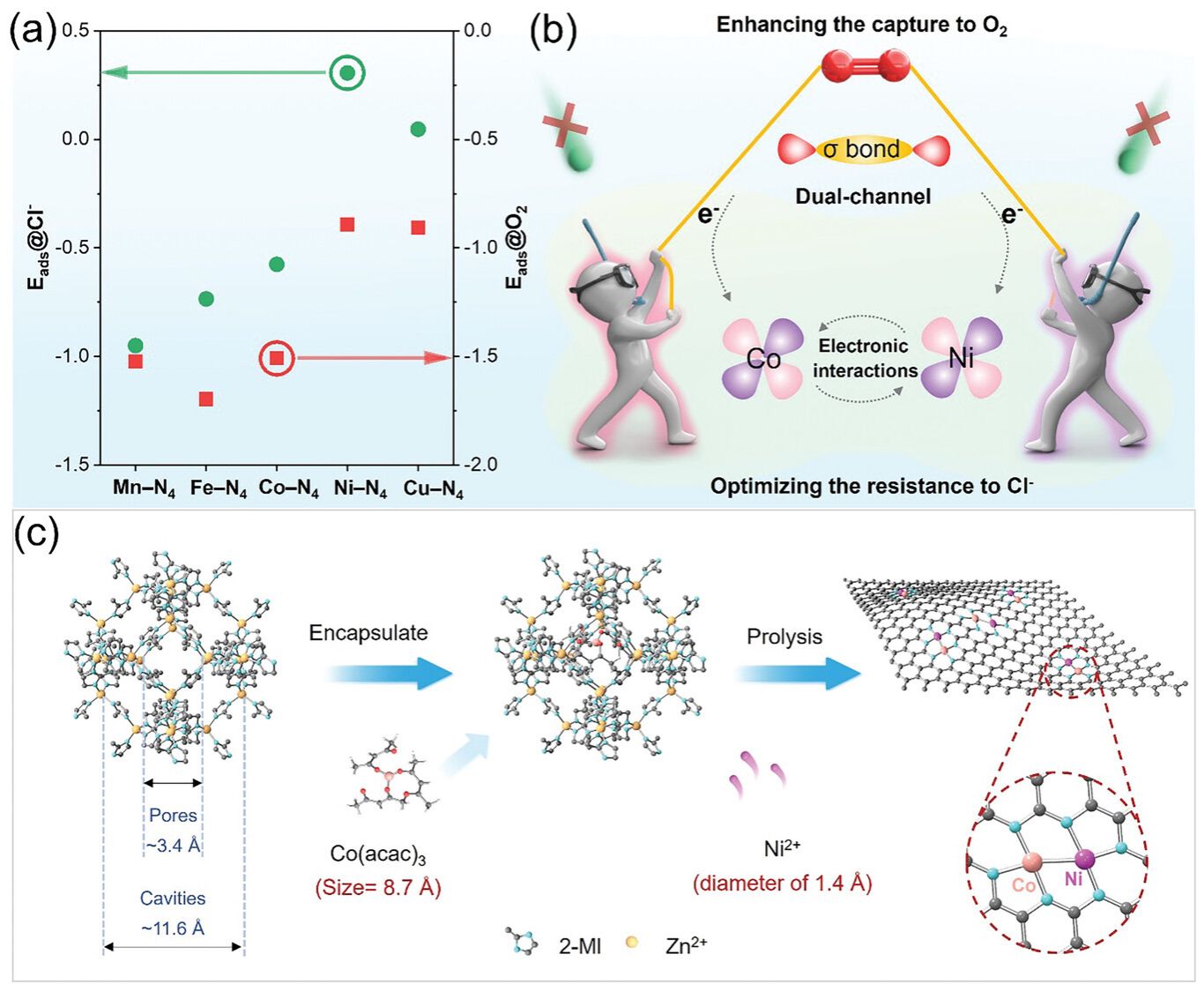
图1. a) O2和Cl−对不同催化位点的Eads的理论计算结果,包括Mn-N4、Fe-N4、Co-N4、Ni-N4和Cu-N4; b) 该催化剂在海水电解质中的ORR催化机理示意图; c) CoNi-DAC的合成工艺说明。
本文要点
要点一:CoNi-DAC表征
通过DFT计算认为双原子CoNi-DAC催化剂可能具有提高天然海水中ORR效率和稳定性的巨大潜力。扫描电子显微镜(SEM)和透射电镜(TEM)显示CoNi-DAC催化剂呈现均匀的菱形十二面体形态,X射线衍射(XRD)表明Co和Ni原子分散在衬底中。拉曼光谱证实了样品具有相似的碳化程度。布鲁诺尔-emmett-Teller(BET)吸附-解吸曲线表明样品存在微孔。能量色散x射线光谱(EDS)映射显示了Co、Ni、N、C元素在基底中的高度均匀分布。高角度环形暗场扫描透射电镜(AC-HAADFSTEM)图像显示了单原子和双金属对共存电子,能量损失谱(EELS)发现Co和Ni形成了相邻的双金属对,证明了双原子催化剂的成功合成。通过XANES图谱和EXAFS光谱得出样品可能结构。

图2. a) TEM图像; b) HAADF-STEM图像; c) EDS映射; d,e) CoNi-DAC催化剂的AC-HAADF-STEM图像; f) 图2e中矩形框中突出显示的双金属对A1-B1的拟合映射; g) 用EELS分析的CoNi-DAC结构; h)在A1-B1和A2-B2区域的两个双原子Co-Ni位点上得到相应的强度分布; i) 观察到的双原子对中的统计Co-Ni距离。
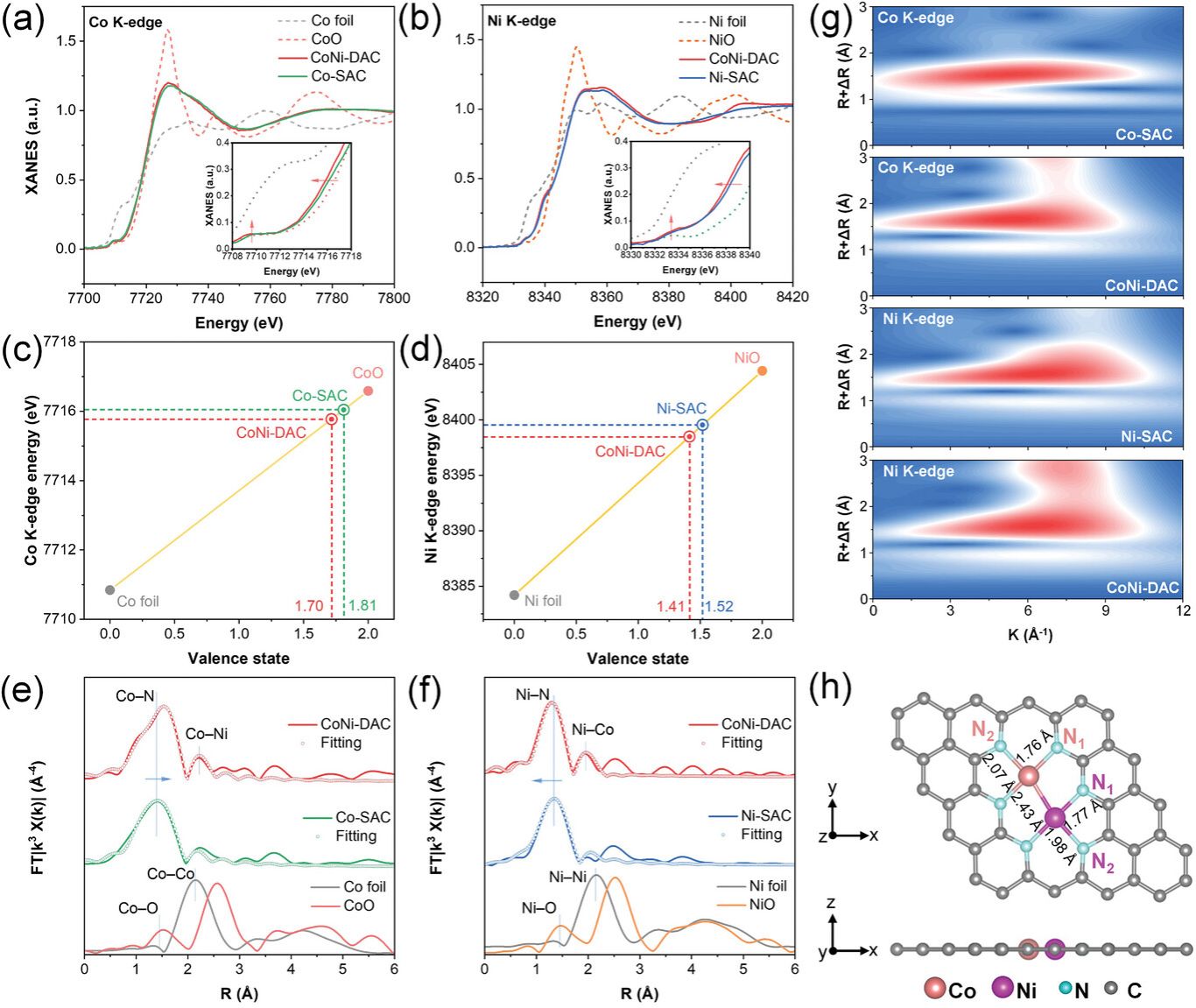
图3. a)Co箔、氧化亚钴、CoNi-DAC和Co-SAC的Co k边缘的XANES光谱。b)Ni箔、一氧化镍、CoNi-DAC和Ni-SAC在Ni k边缘的XANES光谱;通过平均XANES光谱的线性上升边来确定c) Co和d) Ni价态; e) CoNi-DAC和Co-SAC的Co K-边缘和f) CoNi-DAC和Ni-SAC的Ni K-边缘的FT-EXAFS和拟合曲线; g) CoNi-DAC、Co-SAC和Ni-SAC的Co和Ni k-边缘EXAFS的MWT图像; h) CoNi-DAC的优化结构模型。
要点二:电催化ORR性能
CoNi-DAC在天然海水中优越ORR催化动力学,达到了0.79 V的半波电位(E1/2)和在天然海水中近700 h的特殊长期耐久性。电化学双层电容(Cdl)测试进一步证明了其优越的ORR活性。催化剂的Cl−抵抗行为评估实验表明CoNi双金属位点对Cl−的吸附具有免疫力。恢复的极限电流密度和电荷转移电阻表明,CoNi-DAC活性位点没有明显的降解。这些结果突出了CoNi-DAC催化剂显著的电催化活性和稳定性,因为其强大的亲氧性能和对Cl−的抗性,使其成为一种在海水环境中极有前途的ORR电催化剂。
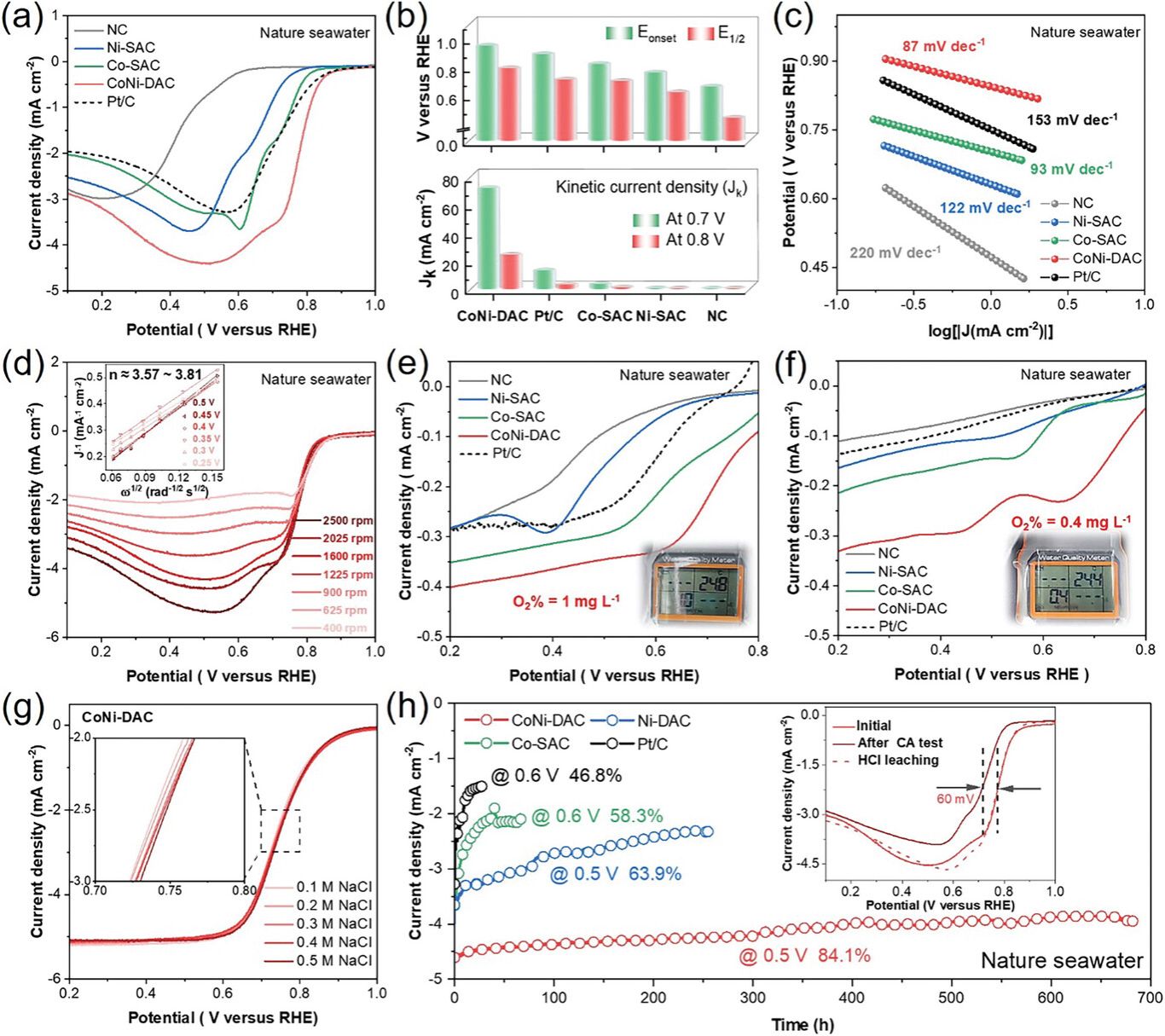
图4. a) CoNi-DAC、Co-SAC、Ni-SAC、NC、Pt/C在5 mV·s−1旋转1600 rpm氧气环境下的ORR LSV极化曲线; b) 不同催化剂在0.7和0.8 V下的起始电位(Eonest)、半波电位(E1/2)和动力学电流密度(Jk)的比较; c) 所有电极对应的Tafel斜率; d) CoNi-DAC在不同转速下的ORR极化曲线,插图显示了的CoNi-DAC基于0.25、0.3、0.35、0.4、0.45和0.5 V的LSV曲线的拟合K-L曲线; 在贫氧条件下, CoNi-DAC、Co-SAC、Ni-SAC、NC和Pt/C的ORR极化曲线e) 1 mg·L−1和f) 0.4 mg·L−1; g) CoNi-DAC在0.1、0.2、0.3、0.4、0.4和0.5 M氯化钠在1600 rpm下的ORR极化曲线; h) CoNi-DAC、Co-DAC、Ni-SAC和Pt/C的CA曲线,插图显示了CoNi-DAC在CA试验前后和酸浸后的ORR极化曲线。
要点三:机理分析
CoNi-DAC的ATR-FTIR光谱表明金属位点之间形成的O2桥(*O-O*),水分子在ORR过程中在催化剂表面积累。反应中间体与应用电势的FTIR高度表明Co-Ni双原子位点的构建可以改变Co-O-O向Co-O-O-Ni中间结构。同时,水分子在CoNi-DAC表面的快速积累增强了O2的活化,跳过了Co-SAC结合途径中*OOH的产生,从而加速了ORR动力学。原位拉曼光谱表明ORR 解离机制的发生是由 Co-Ni双原子活性位点的构建显着诱导的。
氧以桥状结构(*O-O*)吸附在CoNi-DAC中的Co-Ni对位点上。此外,更多的水分子的聚集促进了Co-O-O-Ni构型中O-O键的直接断裂和形成*O的质子化。然后,还原电势驱动桥接氧中间体的裂解,导致在Co位上形成*OH,在Ni位上形成*O。这是因为在孤立Co位上形成*OH的能垒低于在孤立Ni位上的能垒。随后,在Co-Ni对的Co和Ni位点分别发生了稳定的*OH吸附。吸附在Ni位点上的*OH优先解离,因为Co位点与*OH的结合更强。最后,Co位点上的*OH解离。ORR的这一解离途径可以在CoNi-DAC中的Co-Ni双原子位点上实现,从而加速了ORR的反应动力学。
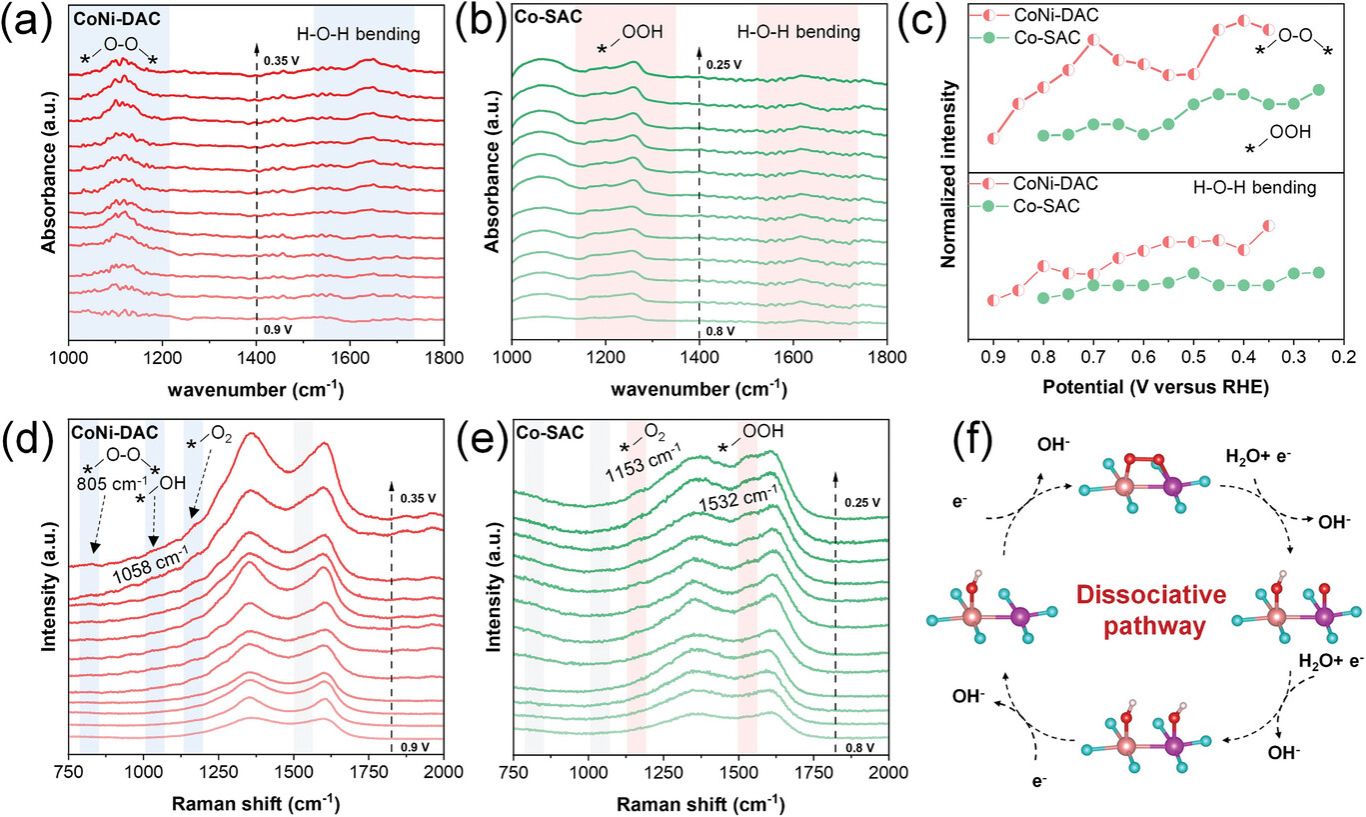
图5. a,b) 在0.5 M氯化钠中,CoNi-DAC和Co-SAC的原位ATR-FTIR光谱; c) *O-O*、*OOH和H-O-H弯曲随外加电位的详细变化; d,e) CoNi-DAC和Co-SAC在0.5 M氯化钠中的原位拉曼光谱; f) CoNi-DAC的反应机理。
要点四:CoNi-DAC作为阴极在SWB中的实际应用
以CoNi-DAC为阴极,以镁合金板为阳极,以天然海水为电解质,组装了一种自制的SWB,以CoNi-DAC为阴极的SWB的峰值功率密度达到3.3W·L−1,超过了商用的Pt/C(0.5W·L−1)。此外,基于CoNi-DAC的SWB在不同的电流密度下始终保持较高的电压,表明了电荷转移动力学较快和较低的电化学极化。SWBs的恒流放电曲线显示,在CoNi-DAC的50 mA L−1电流下,保持1.45 V的放电电压至少500 h,远远高于Pt/C。根据放电过程中金属阳极的质量损失,比容量为1670mAh·g−1。导致放电电压衰减的主要原因是阴极钙沉淀物的沉积和阳极的消耗。因此,该电池的有效和稳定运行凸显了其在基于CoNi-DAC的SWB商业化方面的巨大潜力。
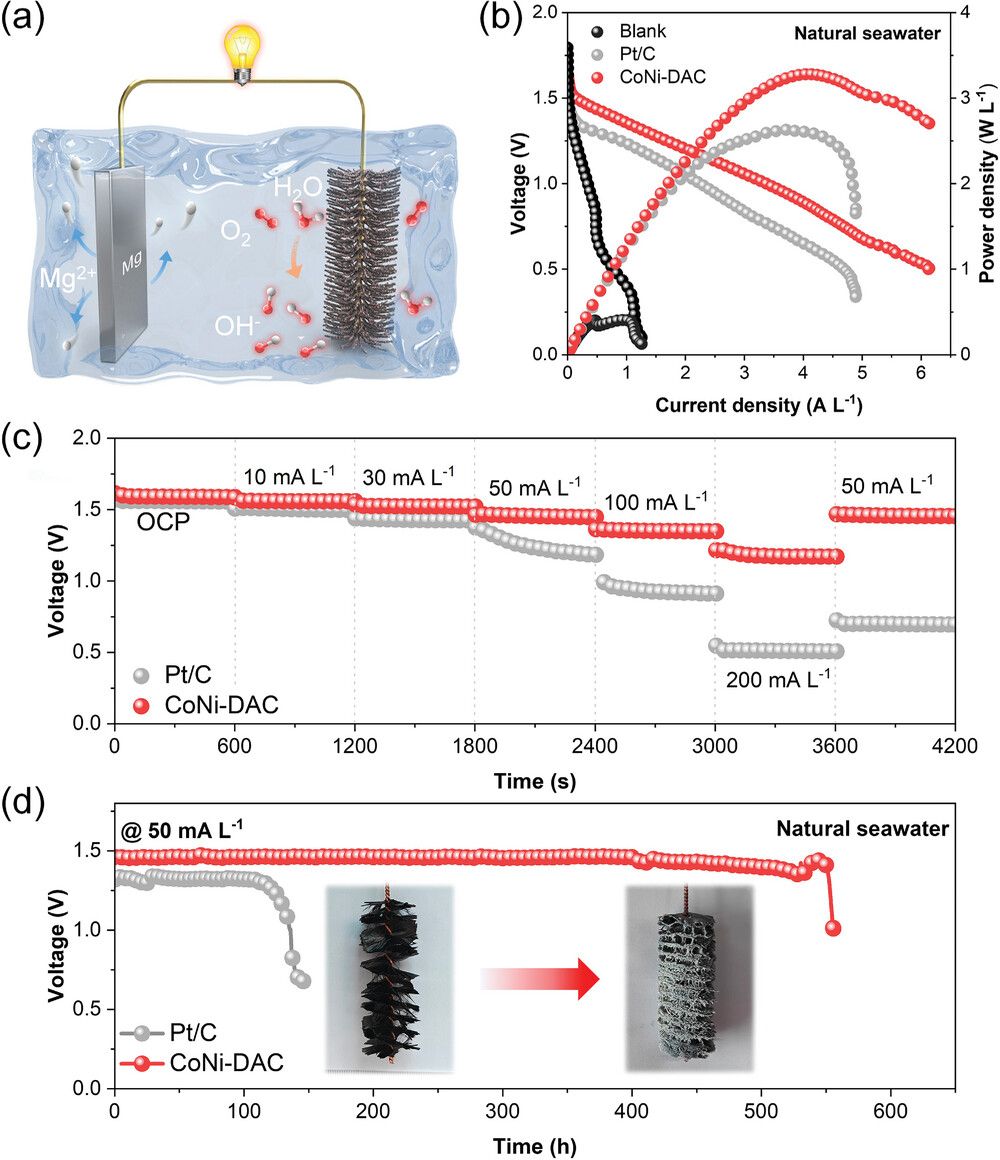
图6. a) SWB的示意图; b) 以CoNi-DAC和Pt/C为阴极催化剂的电池放电极化曲线和计算功率密度; c) 以CoNi-DAC和Pt/C为阴极催化剂,研究电池在不同电流密度下的放电曲线; d) 在电流密度为50 mA L−1时,CoNi-DAC和Pt/C催化下SWB的静电流放电曲线,插图为试验前后阴极的照片。
结论
简而言之,本研究通过主客体策略设计了一种具有相邻Co-Ni双原子位点的CoNi-DAC催化剂,具有高亲氧性和Cl−腐蚀性。实验结果表明,CoNi-DAC在天然海水中显著的ORR性能源于在Co-Ni双原子位点上出现的ORR解离路径。具体来说,i) 低价态相邻双原子位点的存在有利于ORR过程中直接的O-O裂解,从而实现主要的四电子途径,增强其固有的催化活性;ii) Ni-N4位点的Cl−吸收能增强了Cl−耐腐蚀性,从而提高了CoNi-DAC在天然海水中的稳定性。其协同增强机制为ORR电催化剂的合理设计和海水环境中能量转换装置的发展提供了有价值的见解。