海南大学邓意达/郑学荣/王浩志团队ESM(芦俊达、卢琪博士一作):钴原子团簇相互作用协同增强海水中氧还原反应的活性
发布时间:2025-01-23
海南大学邓意达/郑学荣/王浩志团队ESM(芦俊达、卢琪博士一作):钴原子团簇相互作用协同增强海水中氧还原反应的活性

文章链接:https://doi.org/10.1016/j.ensm.2023.103093
第一作者:芦俊达、卢琪
通讯作者:邓意达、郑学荣 、王浩志
通讯单位:海南大学、天津大学
论文DOI:10.1016/j.ensm.2023.103093
全文速览
海水电催化对于各种能量存储和转换系统来说是非常需要的,例如直接使用海水作为电解质的水分解和金属燃料电池。然而,氯离子(Cl−)在正极活性位点上的吸附会恶化氧还原反应(ORR)活性和稳定性,从而降低电池性能。在这项工作中,首先设计了一个电催化剂模型,其中Co原子簇被氮掺杂碳基底上的卫星Co单原子紧密包围,称为Co-ACSAs。理论计算结果表明,Co团簇具有较强的Cl−结合能,可作为Cl−的预吸附基团,充分暴露Co单原子作为ORR活性位点,具有较强的O2吸附能,促进氧还原反应过程。然后,开发了一种超快高温冲击策略,通过控制碳基质中的 N 浓度来合成 Co-ACSAs。受益于Co-ACSAs中Co团簇和卫星Co单原子之间适中的相互作用距离,通过互连六原子环的调制优化了Co团簇和Co单原子的电子结构。因此,Co-ACSAs表现出优异的 ORR 活性和耐久性,起始电位和半波电位分别为 0.885 V 和0.782 V,并在海水电解质中连续催化 485 小时。基于Co-ACSAs的海水电池在5 mA cm−2下的放电电压为1.41 V,并实现了超过390小时的稳定供电。
背景介绍
从自然资源中获取能源具有重大意义。海水是最丰富的自然资源,覆盖地球表面的70%。海水电催化技术,例如全水分解和金属燃料电池,为直接将海水转化为氢气和电力为海上工作设备提供动力提供了巨大的潜力。目前,大多数海水利用技术都是电化学装置,其中阴极电催化剂的氧还原反应(ORR)活性和稳定性是决定储能和转换电池系统整体性能的核心。然而,与传统碱性电解液中的电池不同,海水中氯离子(Cl−)的存在会严重降低阴极电催化剂的ORR活性和耐久性。Cl−在电催化剂表面催化位点上的吸附会毒害活性位点,从而抑制氧分子的吸附,阻碍O-O键的断裂,诱导ORR途径从四电子向二电子转移。因此,在恶劣的海水环境下,电催化剂需要满足以下要求:(i)催化活性位点与Cl−无相互作用或吸附能较低;(ii)催化剂应具有较大的活性位点暴露量,以增加其与活性物质的接触,例如质子、电子和氧中间体;(iii)活性位点应具有最佳的电子和化学键结构,以提高催化活性和稳定性。
铂基 ORR 电催化剂通常表现出高 ORR 效率。但它们成本较高,并且在海水电解液中很容易被腐蚀。尽管开发了涂层和缺陷工程策略,但其稳定性仍然达不到工业要求。最近,单原子(SA)催化剂,主要是金属氮-碳(M-N-C)系列,由于其原子利用率高、电子结构可调、每个活性位点具有优异的内在催化活性,对于ORR过程非常有效。此外,单原子电催化剂的合理设计将使其具有优异的耐氯中毒性能,使其有望在海水电解液中得到应用。例如,通过微波加热方法合成了锚定在石墨烯/CNT基底上的Fe-SAs(Fe-N-G/CNT),并在海水中显示出704 mV的ORR半波电位,在10 mA cm-2下放电电压为1.18 V。海水金属空气电池(SWB)。然而,SA的活性和稳定性对于ORR 过程是权衡关系。平衡活性和稳定性的常见策略是通过掺杂杂原子或构造缺陷来调节金属位点与基底之间的电子调节,但调节范围较窄且难以打破活性与稳定性的权衡关系。此外,由于SA中金属原子分布稀疏,这些金属原子之间的电子相互作用通常可以忽略不计。最近报道的结果表明,通过引入相邻原子团簇(AC)进一步调节单原子的电子结构可能会提供增强其内在催化活性和稳定性的机会。AC 可以提供具有相邻金属原子的活性位点,同时将它们保持为单独的位点。AC和SA之间潜在的协同相互作用产生了独特的电子特性,有望提高电催化性能。然而,由于其高表面能且易于团聚成纳米颗粒,在导电基底上构建稳定的ACs调制SA(ACSA)仍然是一个巨大的挑战。此外,很少有人关注ACSAs在海水电解质中的ORR机制,这可能为设计活性稳定的SWBs电催化剂提供新的途径。
本文亮点
该项工作设计的电催化剂 Co-ACSAs,经理论计算结果表明Co-ACSAs中的CoAC表现出更强的Cl−吸附能但更低的O2吸附能。计算结果表明,Co-ACSAs中的Co-ACs在保护Co-SAs免受Cl−中毒方面发挥着关键作用,并且Co-SAs位点上较强的O2吸附能将促进海水电解质中的ORR过程。受主要理论计算的启发,开发了一种超快高温冲击 (HTS) 策略,用于一步将 Co-AC 和卫星 Co-N4单原子锚定在氮掺杂碳基底 (Co-ACSAs) 上。实验和理论结果表明,受益于Co-ACs和Co-SAs之间的最佳距离,Co-ACs可以通过互连的六元环调节附近Co-SAs的电子结构,从而促进氧活性物种的吸附Co-SAs 并通过降低相应的能垒来加速 OOH* 的形成。 Co-ACSAs 中 Co-ACs 和 Co-SA 之间的强烈调制产生了令人印象深刻的 ORR 活性和在海水电解质中的长期稳定性,优于Co-SA 和 Co 纳米颗粒 (Co-NP) 的纯相。
图文解析
要点1:首先建立了一个原子电催化剂模型,其中Co-ACs被Co-ACSAs中的卫星Co-SA紧密包围(图1a)。还建立了碳基底上 Co-SA 的纯相模式以进行比较。进行密度泛函理论 (DFT) 计算来计算 Cl− 和 O2 分子在所有潜在催化位点上的吸附能,包括 Co-ACSAs 模型中的 Co-AC 和 Co-SA 位点,以及孤立的 Co Co-SA 纯相中的N4位点(图 1b)。据报道,碳基质中的N位点通常充当稳定过渡金属原子的锚定位点(图1c)。因此,我们将N的质量比控制在1wt%、3wt%和5wt%的比例,然后Co原子可以在衬底上几何分离,并分别形成Co NP、Co ACSA和Co SA。此外,这种合成方法被发展成为合成Fe-ACSAs和Ni-ACSAs的通用策略。HAADF-STEM图像(图1d-e)显示原子团簇被单原子紧密包围,并且团簇和单原子之间的距离小于1.0 nm,这为团簇和单个原子之间的电子调制提供了极好的机会。元素映射图像显示Co-ACSA均匀分布在衬底上(图1e)。
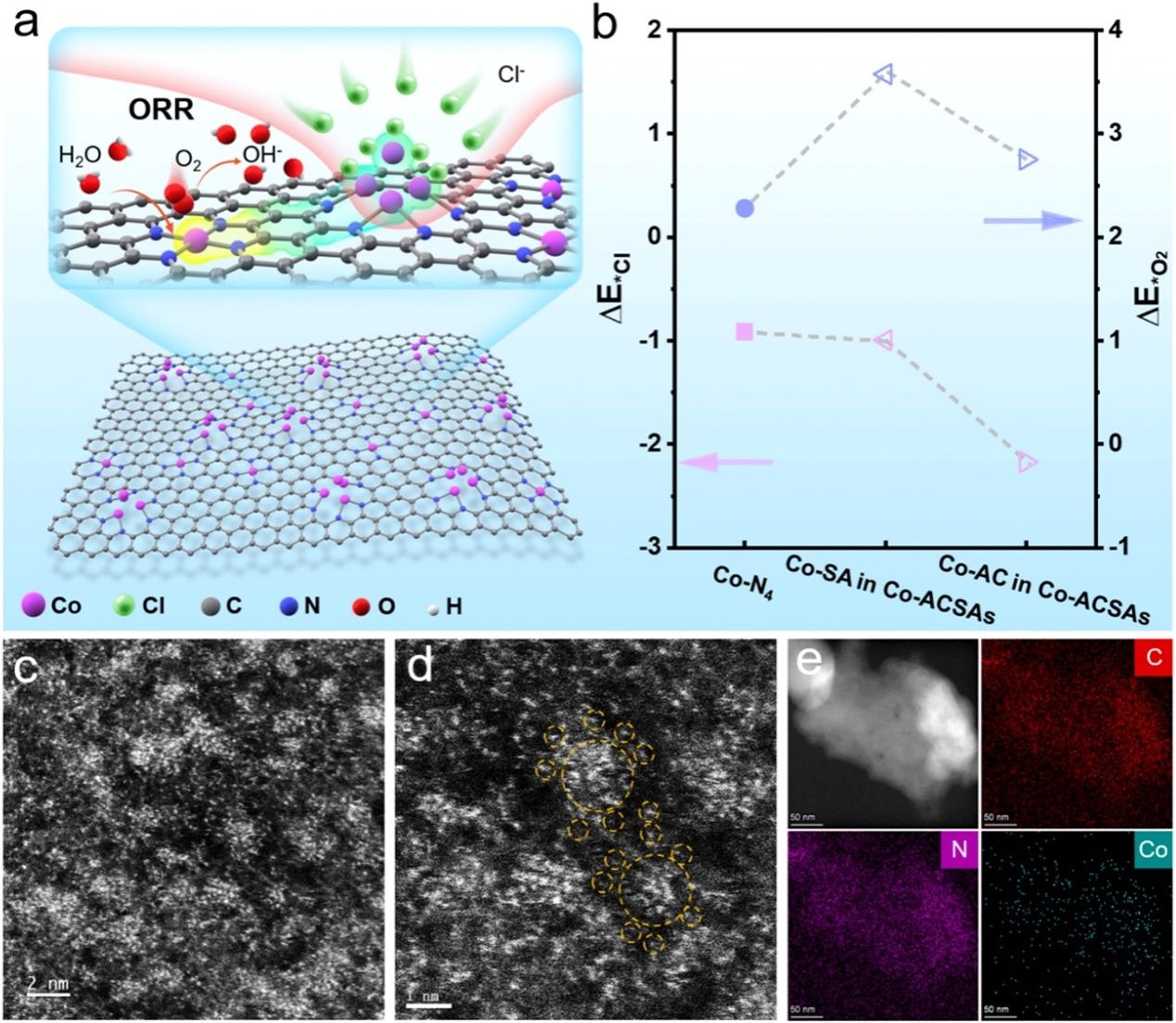
图1.(a) Co-ACSAs 在海水电解质中所提出的 ORR 催化机制的示意图。 (b) Cl−和O2分子对Co-ACSAs和Co单原子不同活性位点的吸附能(ΔEads)的理论计算结果。 (c-d) Co-ACSAs 的 HAADF-STEM 图像和 (e) 元素映射图像。
要点2:图2a显示了Co-ACSAs、Co-SAs和参考样品(Co箔和CoO)中Co K边缘的X射线吸收近边缘结构光谱(XANES)。为了定量评估催化剂中Co的价态,我们选择上升K边缘的近线性部分进行积分,并将积分平均强度定义为K边缘位置。Co-ACSAs和Co-SAs中Co的吸收阈值位置接近CoO的吸收阈值位置而远离Co箔,这意味着两种催化剂中Co的二价态。随后,根据K边位置定量计算催化剂中Co的价态,对于Co-ACSAs和Co-SAs分别为Co1.68+和Co1.73+(图2b)。原子级催化剂中Co的氧化应该是由Co-N-C配体的形成引起的,其中电子被Co吸引到N-C。与Co-SAs相比,Co-ACSAs中Co的价态较低应该是由于Co团簇的存在,其中部分Co不能与N-C底物杂化,从而在团簇中形成Co-Co键在 Co-ACSA 中。o-ACSA 和 Co-SA 中 Co-K 边缘的扩展 X 射线吸收精细结构 (FT-EXAFS) 光谱的 k3 加权傅立叶变换如图 2c 所示。在 Co-ACSA 和 Co-SA 中都可以观察到 R 空间中约 1.4 Å 处的 Co-N 峰。然而,仅在 Co-ACSA 中观察到 2.2 Å 处的 Co-Co 散射峰,证明 Co-ACSA 中 Co 团簇和单个原子共存。弱的Co-Co散射峰表明Co团簇的粒径非常小,应该被定义为原子团簇。o箔、Co-ACSA和Co-SAs中Co的k3加权EXAFS光谱的MWT如图2d-f所示。在图 2d 中,MWT 等值线图显示 k 空间中约 7.0 Å−1处的强度最大值,这可以归因于 Co 箔中的 Co-Co 散射路径。与Co-SAs的MWT图像相比,Co-N和Co-Co最大值分别出现在Co-ACSAs中4.2 Å−1和7.0 Å−1处(图2e-f),这与Co-ACSAs的MWT图像一致。
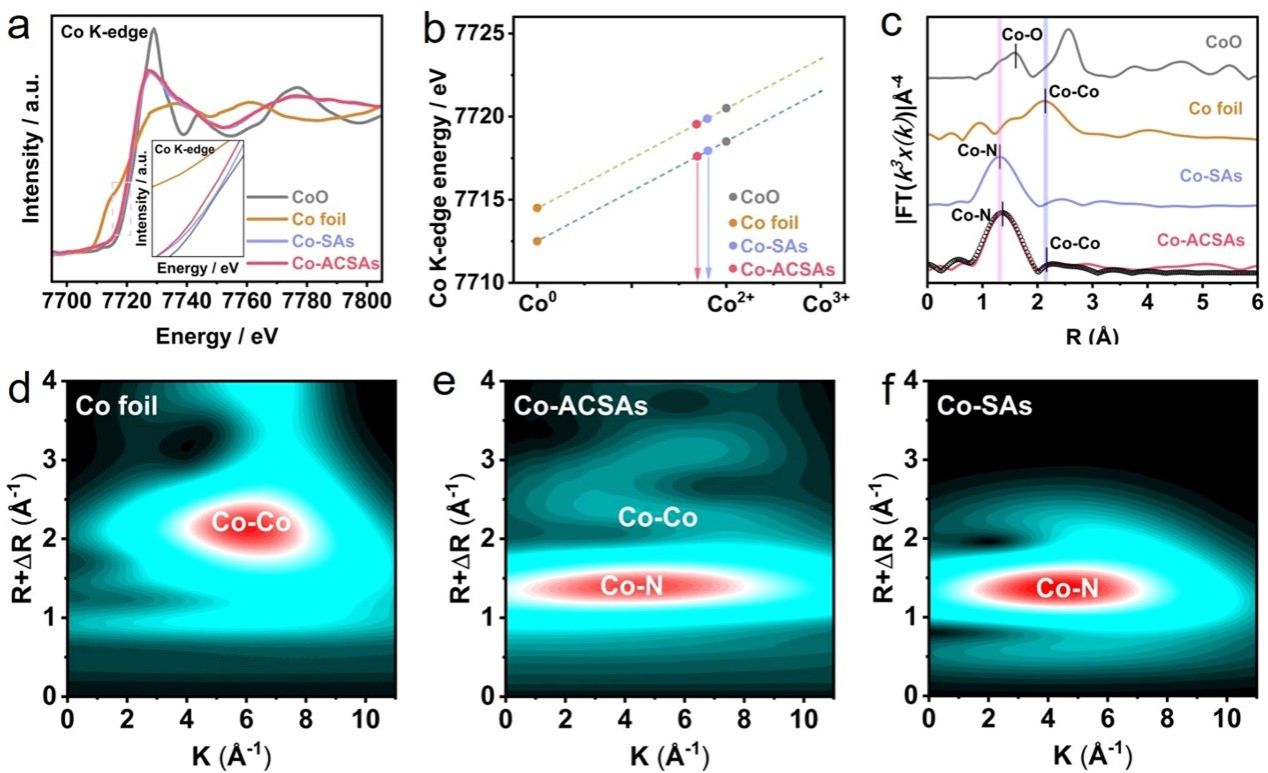
图2. (a) Co-ACSAs 和 Co-SA 的 Co K 边缘 XANES 以及 Co 箔和 CoO 的参考光谱。 (b) Co-ACSAs 和 Co-SA s中计算的 Co K 边缘位置和 Co 价态。 (c) Co箔、Co-ACSAs和 Co-SAs 中 Co K 边缘的k3 加权 EXAFS 光谱的 FT 光谱。 (d-f) Co 箔、Co-ACSAs 和 Co-SAs 的 MWT 图像。
要点3:LSV 结果表明,Co-ACSAs 显示出令人印象深刻的半波和起始ORR过电势,E0=0.885 V 和 E1/2=0.782 V,远高于Co -SA、CoNP和Pt/C。商业Pt/C的LSV曲线没有表现出明显的峰值和有限的电流密度,这可能是由海水中严重有毒的Cl−引起的。N-C在天然海水中表现出弱的ORR活性。相应地,Co-ACSAs 的塔菲尔斜率也比 Co-SAs 、Co-NPs和Pt/C更小,表明其在天然海水中具有优越的ORR催化动力学。通过测试不同转速下的LSV曲线,这表明Co-ACSAs具有最高的反应动力学,Co-ACSAs 在天然海水中表现出显着的ORR 稳定性,Co-ACSAs 在0.65 V vs. RHE 下表现出最小的降解率。如图3e-f所示,在含Cl−混合电解液中测试时,Co-ACSAs的极限电流密度下降较小。然而,Co-SAs和Co-NPs在相同条件下表现出巨大的下降,表明Co-SAs和Co-NPs中更多的活性位点被Cl−毒害,但对Co-ACSAs 的活性位点暴露。图3g-h 所示,与KOH电解液中的测试结果相比,在含 Cl−电解液中测试的 Co-ACSAs 的ECSA 值下降了,远小于KOH 电解液中的测试结果。
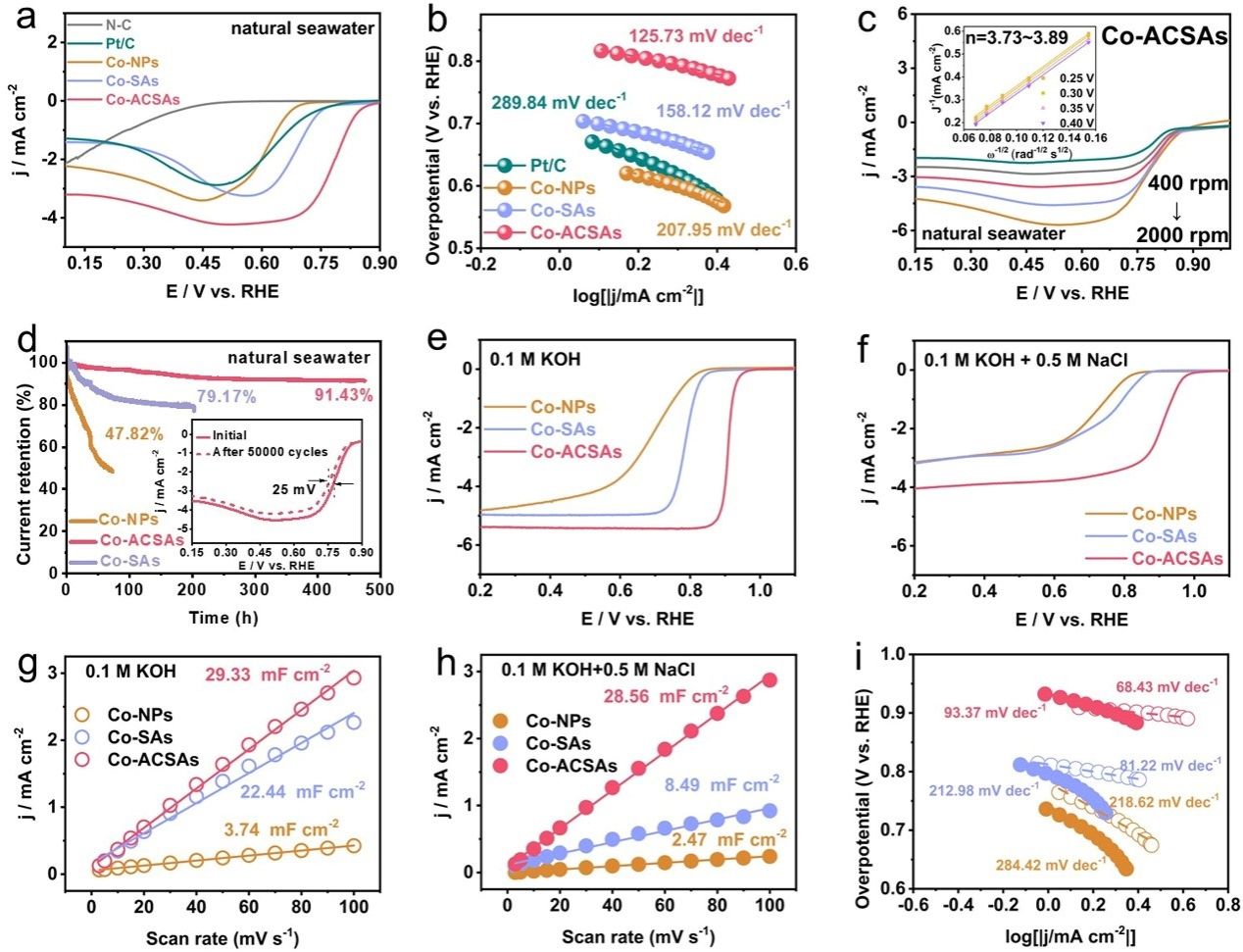
图3.(a) Co-SAs、Co-ACSAs 和 Co-NPs 在 O2 饱和天然海水中在 5 mV s −1 下、转速为 1600 rpm 时的 ORR LSV 极化曲线。 (b) 塔菲尔斜坡。 (c) Co-ACSAs 在不同转速下的 ORR 极化曲线,插图显示了基于 0.25、0.30、0.35 和 0.4 V 的 LSV 曲线的 Co-ACSA 拟合 K-L 图。(d) Co-ACSAs 的计时电流曲线NP、Co-ACSAs 和 Co-SA 在 0.65 V vs RHE 下,插图显示 Co-ACSAs 在 50,000 个CV 循环之前和之后的 ORR 极化曲线。 Co-SAs、Co-ACSA 和 Co-NPs 在 O2 饱和 (e) 0.1 M KOH 和 (f) 0.1 M KOH + 0.5 M 中在 5 mV s-1 下、转速为 1600 rpm 时的 ORR LSV 极化曲线氯化钠。所制备的钴基催化剂在 (g) 0.1 M KOH 和 (h) 0.1 M KOH + 0.5 M NaCl 中的 Cdl 值。 (i) 相应的塔菲尔斜率。
要点4:通过DFT计算揭示了Co-ACSAs中原子团簇和单个原子之间促进海水电解质中ORR活性的协同机制。原子团簇的几何尺寸以及原子团簇与单个原子之间的距离等关键因素可能对Co-ACSAs中两种不同Co物种之间的相互作用产生潜在影响,我们在理论计算中系统地研究了这些因素。如图4a-d所示。对于距离为2.31 Å 的模型,Co-SA会直接与 Co-AC(Co4)建立Co-Co 配体,因此倾向于形成Co5簇,导致 RDS 的ΔG值更高,为0.7828 eV。对于距离为4.85 Å的模型,Co-ACs和Co-SAs之间会发生强烈的电子调制,导致活性位点周围电子严重积累,并且确定步骤的能垒更大,为0.4829 eV。受益于Co-ACs和Co-SAs之间通过六原子环进行的适度电子调制,Co-ACs和Co-SAs都具有优化的电子分布密度,这使得ORR确定步骤的能垒更小,为0.1998 eV。上述计算结果表明,Co-ACs可以作为Cl−的预吸附位点,并保护相邻的Co单原子免受Cl−的毒害,从而使Co单原子成为ORR过程的真正催化位点。
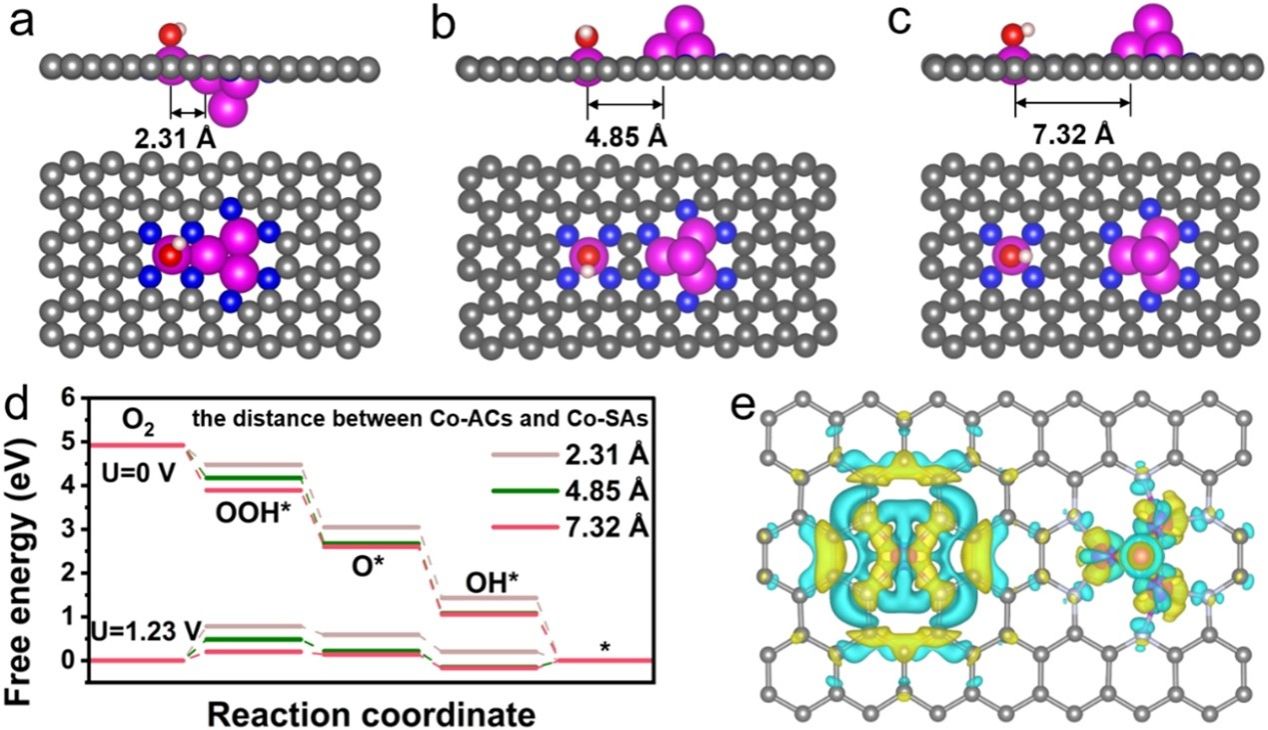
图4.Co-ACSAs 的模型结构(Co:粉红色,N:蓝色和 C:灰色),距离为 (a) 2.31 Å、(b) 4.85 Å 和 (c) 7.32 Å。 (d)计算出具有不同团簇和单个原子之间距离的Co-ACSAs的ORR过程的自由能图。 (e) Co-ACSAs 的电荷密度差异。 (f) Co-ACSAs 中 Co-N4 位点和 Co4 位点以及 Co 单原子纯相上的 Cl− 和 O2 分子吸附能(Cl−:绿色,O2:红色)。
要点5:在测试系统中,测试期间天然海水在O2饱和度下循环。放电极化和相应的功率密度曲线如图5b所示。使用Co-ACSAs作为阴极的SWB的峰值功率密度达到96 mW cm−2,此外,SWBs在1 mA cm−2下的恒电流放电曲线表明,Co-ACSAs可以将放电电压维持在1.41 V至少240 h,这比Co-NPs、Co-SAs和Pt/ C(图5c)。此外,基于Co-ACSA的SWB的开路电压可以达到1.72 V并保持稳定至少6000 s(图5d)。 如图5e所示,基于Co-ACSA的SWB可以保持稳定,以提供频繁变化的1、5和10 mA cm−2的可变电流密度。据了解,基于Co-ACSA的SWB的放电电压、比容量和耐久性均创下了SWB领域的新纪录。为了推动SWB的实际应用,将两个自制的SWB串联起来,可以很好地为发光二极管(LED)供电至少24小时。组装后的SWB器件可以提供5 mA cm−2(1.45 V)的电流密度,并使LED灯稳定工作至少390小时,放电容量为2203 mAh g−1 Mg。
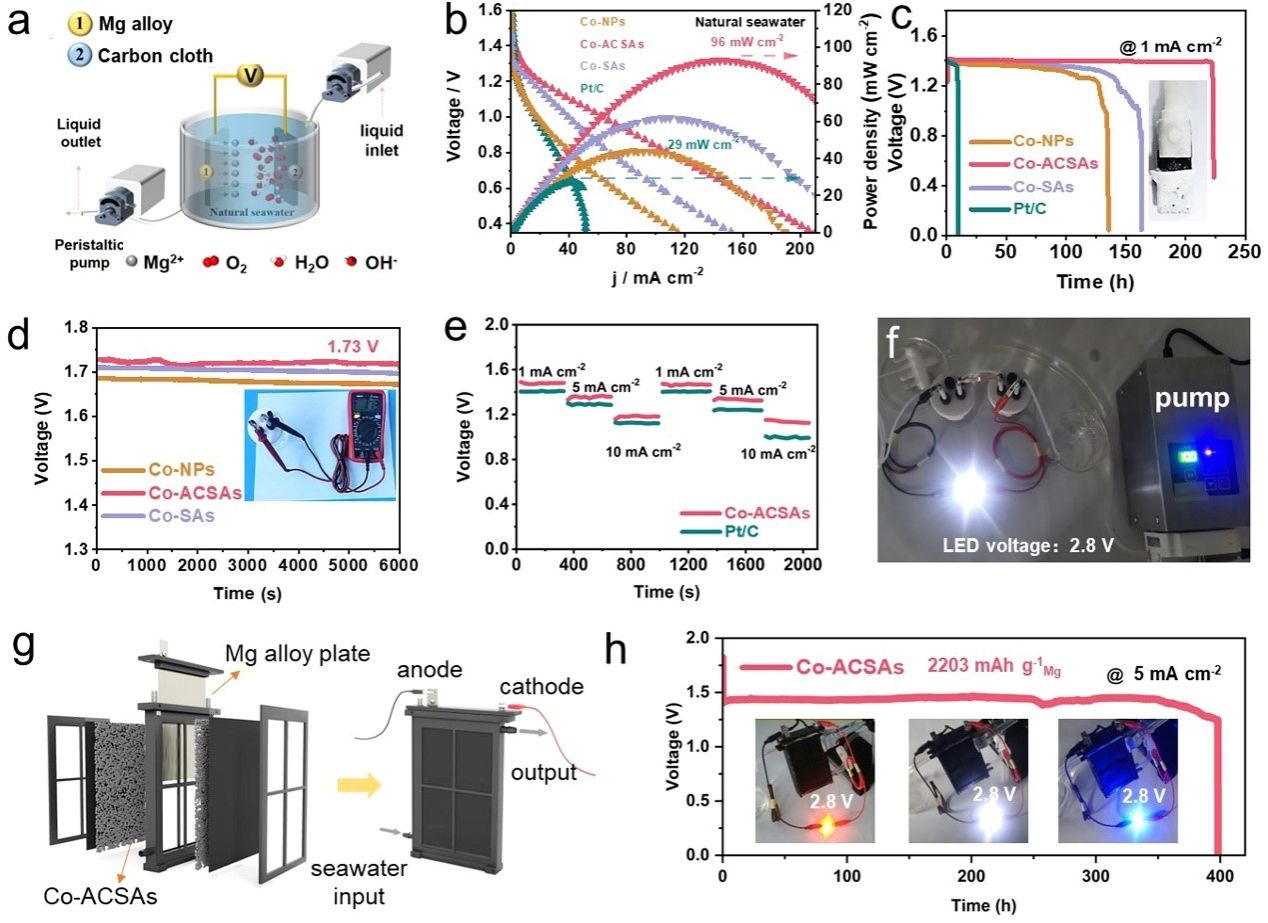
图5.(a) SWB 示意图。(b)使用Co-NPs、Co-ACSA、Co-SAs和Pt/C作为空气电极的电池的放电极化曲线和计算的功率密度。(c) Co-NPs、Co-ACSA、Co-SAs 和 Pt/C 催化的 SWB 在电流密度为 1 mA cm−2 下的恒电流放电曲线,插图为测试后阴极的照片。(d) 使用 Co-NP、Co-ACSA 和 Co-SA 作为空气电极的 SWB 的开路图。 (e) 使用Co-ACSAs和Pt/C作为空气电极的电池在不同电流密度下的放电曲线。(f) 由两个串联的基于 Co-ACSAs 的自制 SWB 点亮的 LED 的数码照片。(g) 定制SWB的示意图。(h) 基于 Co-ACSAs 的接近商用 SWB 在 5 mA cm−2 电流密度下的放电曲线,插图是由两个串联电池点亮的 LED (2.8 V) 的照片。
总结与展望
总之,该项研究开发了一种超快合成原子钴基电催化剂的方法。通过调节碳基质中的氮质量比,可以精确控制从纳米颗粒到原子簇,再到单个原子的催化剂颗粒尺寸。与Co-SAs和Co-NPs相比,Co-ACSA在天然海水中表现出优异的ORR活性和耐久性。本研究中Co-ACSAs的协同增强为ORR电催化剂的合理设计提供了有价值的指导,计算结果表明CoACSAs卓越的ORR性能应受益于以下因素:(1)单催化剂之间的电子调制Co-ACSAs中通过六原子环的原子和原子簇优化了Co-SAs的电子结构,从而增强了其固有的催化活性;(2)原子团簇较强的Cl−吸收能,保护了单个原子免受Cl−中毒,从而保证了大的活性位点暴露; (3)单原子和团簇之间的独立存在协同增强了Co-ACSAs的稳定性。基于Co-ACSAs的SWB具有出色的长时间放电功率密度和稳定性,主要应用于大型光照明设备。本研究中Co-ACSAs的协同增强为ORR电催化剂的合理设计以开发海水环境中的能量转换装置提供了有价值的指导。